What are web services?
Web services are protocols for computers to exchange information over the web (see wikipedia: web service), typically via an API. In biology, web services are becoming more and more common. You can obtain, for example, phylogenetic trees from Open Tree of Life, geographic occurrences from GBIF, and trait data from Encyclopedia of Life, all using web services.
We have a number of pre-made functions in Arbor that can obtain data through web services. These functions can be included in workflows that make it easy to combine your data with data obtained over the web and carry out analyses. In this tutorial we will illustrate one of these applications, starting with a list of species and obtaining a phylogenetic tree from Open Tree of Life.
Example: from a list of species to a phylogenetic tree
We are going to start with a list of species. You can obtain this list as a csv file here; download and save this file somewhere to your computer.
Now, open an Arbor instance (e.g. Arbor web app). You will now need to load in your species list. You can do this by dragging the file that you just downloaded onto the “Browse or drop files” box:
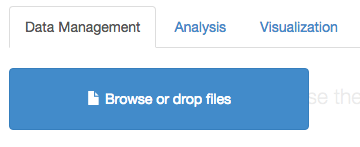
(you can also see our tutorial on using the Arbor webapp) for more information on loading data into Arbor)
We will be building a workflow to do this analysis. Note that you need to be logged in and have write access to a collection to make the next parts work!
Click over to the “Analysis” tab, and create a new workflow. To do that, type the name of the new workflow it the box under “Create new analysis.” Let’s call our workflow “getOTLTreeAndPlot.”
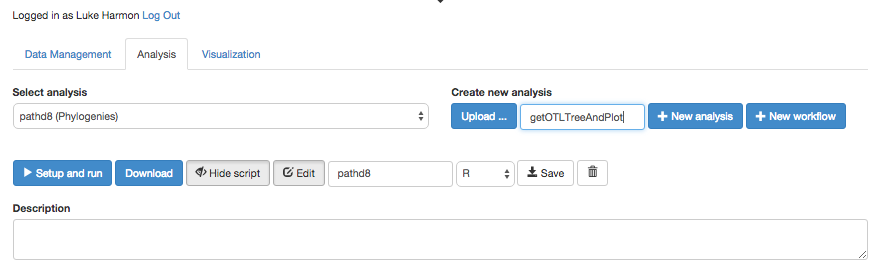
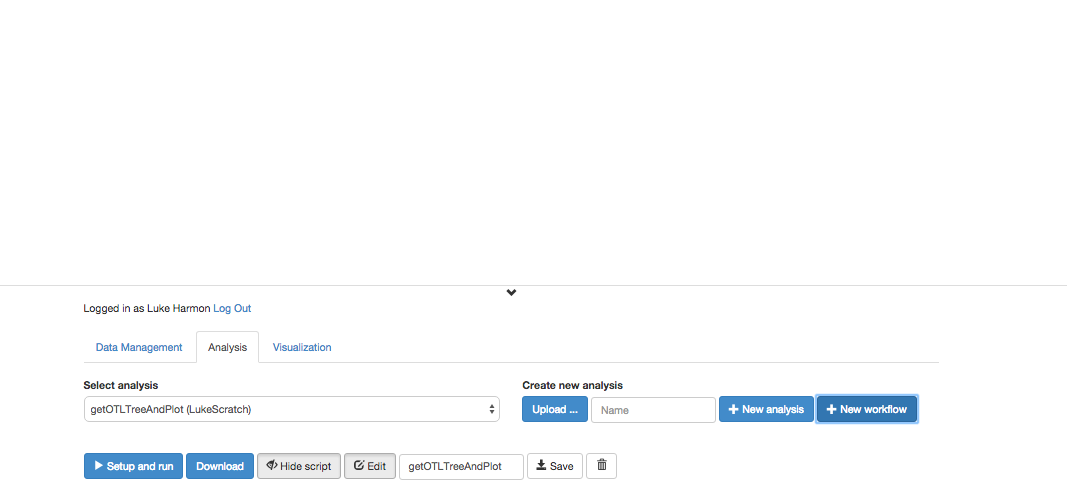
Once you have created the workflow, you should be able to view and edit (but right now it is just a blank white space!)
The first step in this workflow is to match the species names with the names in the Open Tree of Life Taxonomy (OTT). To do this, we will use a function in the “advanced-opentree” collection called “Lookup Names Using OpenTree Taxonomy.” Add this function to your workflow using the “+ add to workflow” button.
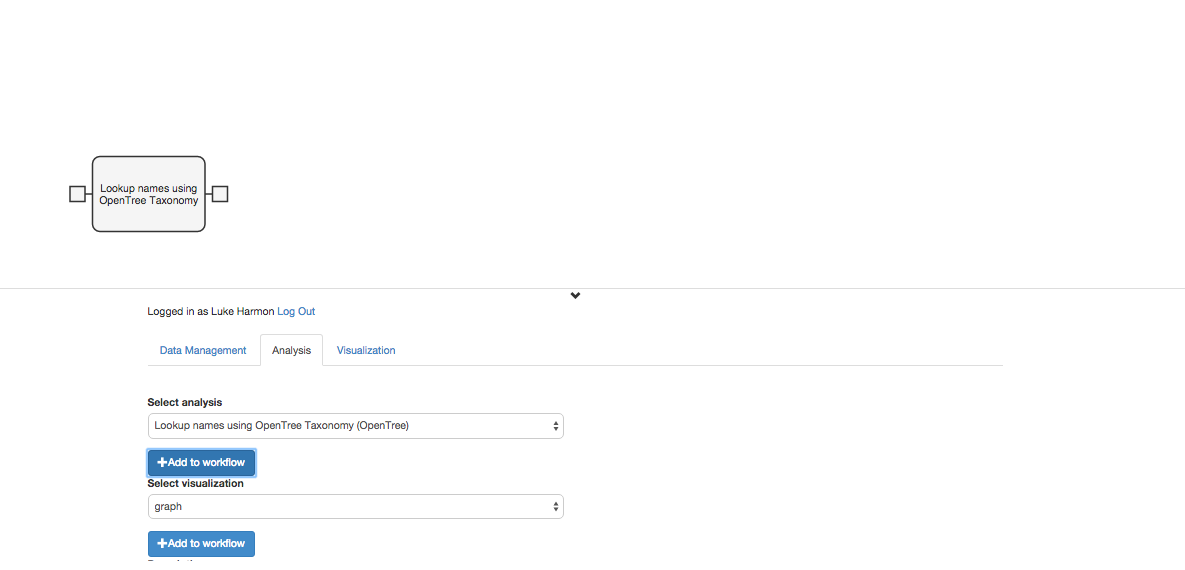
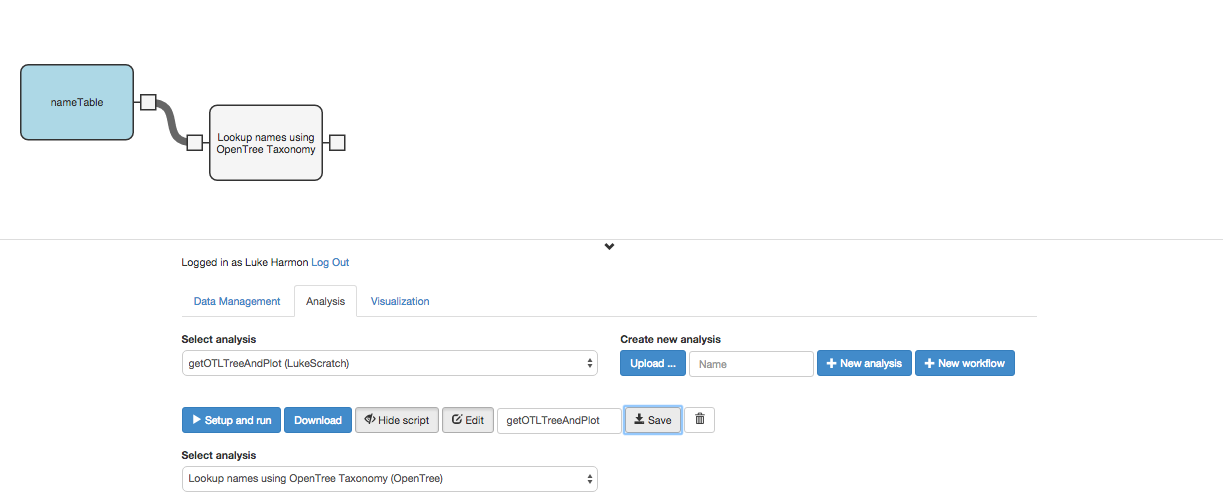
We can click the tab on the left side of that new workflow step to indicate that the user will send a data table directly to this function. So our workflow is now:
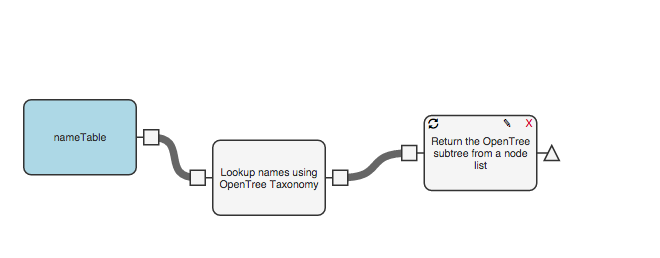
Now we need to pull out an “induced subtree” from open tree of life using the OTT ids that we obtained in the previous step. To do that, add a new function to your workflow, again from the “advanced-opentree” collection: “Return the Open Tree Subtree from a node list.” Add and connect that next step to your workflow:
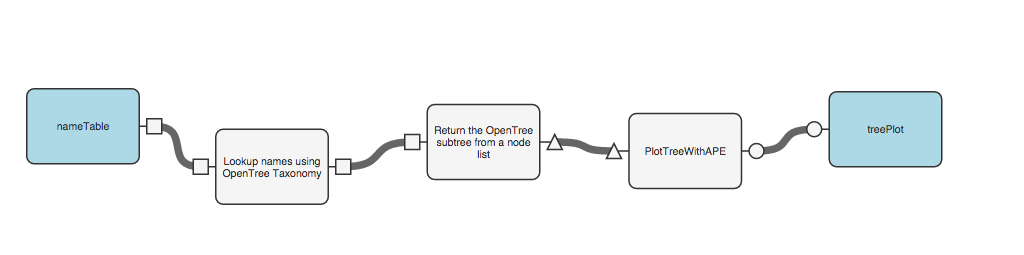
Now we just need to plot our resulting tree. You can use a function from the “advanced-phylogenyTools” collection called “PlotTreeWithApe.” Add that function, connect it to your workflow, and specify that the last output be sent to the user:
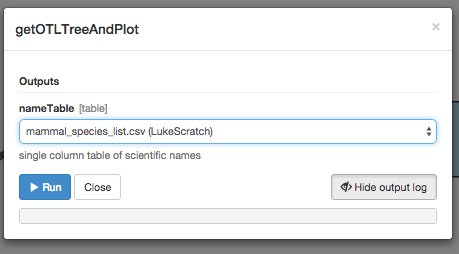
Now just click “Setup and run.” In the box that appears, choose your input file with the list of mammal species names.
The workflow will run for a while:
And then finish:
You can then change to the “visualization” tab, and select the output from your workflow.
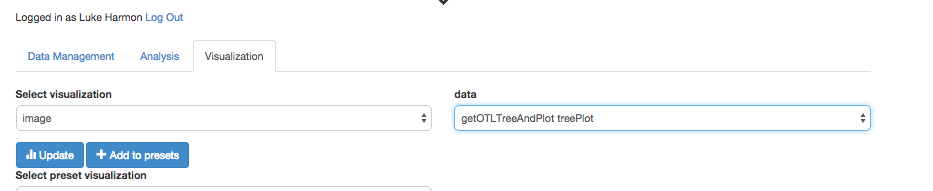
We then see the final result:
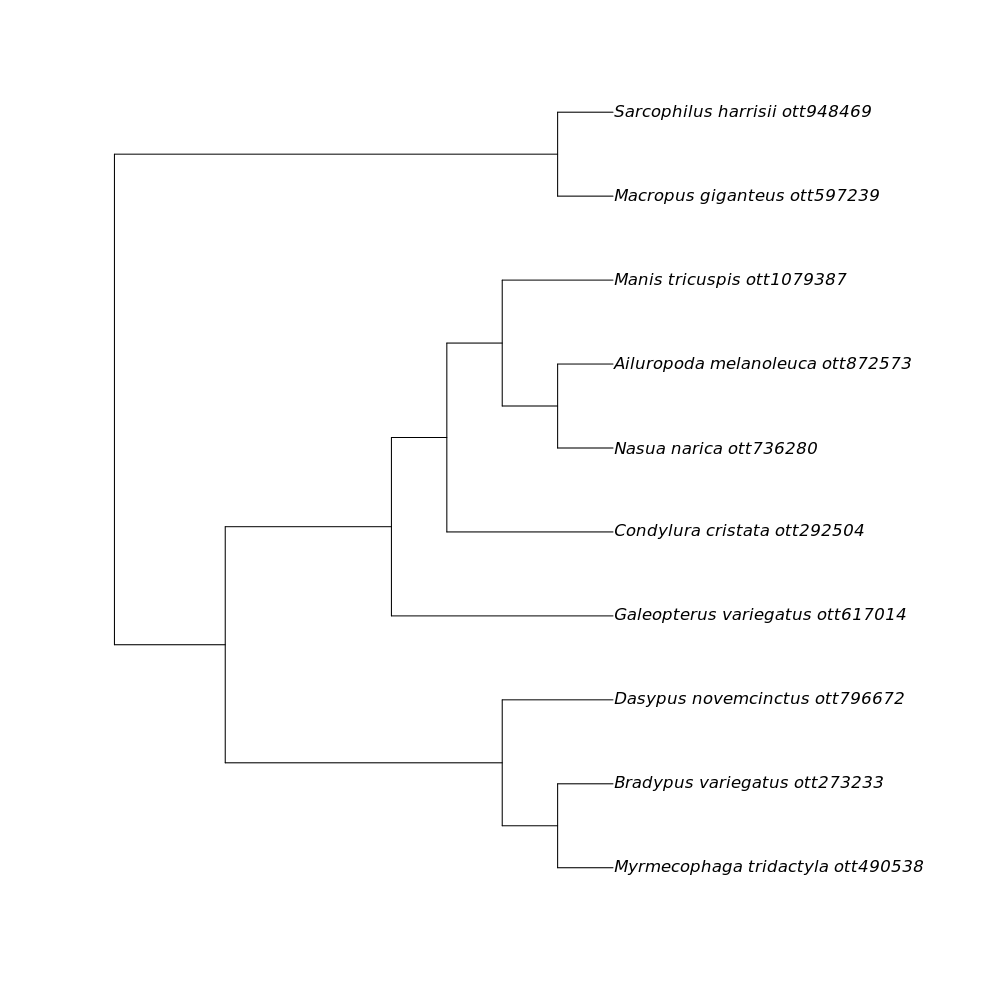
This is a phylogenetic tree pulled from the Open Tree of Life synthesis that includes all of the species in the table that you supplied.